Cover Art Collection






Research Focus 1: Gel-based Proteomics
In-depth proteoform analysis using top-down mass spectrometry enabled by gel-based high-resolution prefractionation of crude protein extracts
Detailed sequence information from intact proteins can be directly acquired via top-down mass spectrometry of proteins, when it is combined with ultra-high-resolution mass spectrometry and powerful molecular fragmentation techniques. In top-down proteomics, or large-scale analysis of protein components extracted from biological samples by top-down mass spectrometry, fractionating crude protein extracts prior to mass spectrometry is essential in detecting trace protein components. Polyacrylamide gel electrophoresis, which enables high-resolution separation of proteome prior to top-down mass spectrometry, is a potentially promising technique. However, the lack of an effective approach to recover the separated proteins from the gel after electrophoresis has prevented its application in top-down proteome analyses. In this study, we developed a novel method for rapid and efficient extraction of intact proteins from polyacrylamide gels. This method allows efficient recovery of intact proteins from a wide range of molecular weight regions after high-resolution protein separation. The establishment of a gel-based top-down mass spectrometry workflow will accelerate the analysis of in-depth top-down proteomics.

Takemori A. et al., PEPPI-MS: gel-based sample pre-fractionation for deep top-down and middle-down proteomics Nat. Protocols. 2025
Takemori A. et al., Size-Based Proteome Fractionation through Polyacrylamide Gel Electrophoresis Combined with LC–FAIMS–MS for In-Depth Top-Down Proteomics. Anal. Chem. 2022
Takemori A. et al., Bottom-up/cross-linking mass spectrometry via simplified sample processing on anion-exchange solid-phase extraction spin column. Chem Comm. 2022
Takemori A. et al., PEPPI-MS: Polyacrylamide-Gel-Based Prefractionation for Analysis of Intact Proteoforms and Protein Complexes by Mass Spectrometry. J Proteome Res. 2020
Development of a novel protein digestion method using BAC gel (dissolvable polyacrylamide gel) and its application to mass spectrometry-based proteomics
Sample pretreatment, which includes removal of contaminants and recovery of the samples from the storage media, for mass spectrometric analysis is crucial to achieve reproducible and accurate results. The goal of this study was to develop an in-gel digestion method to enhance peptide recovery and reproducibility of the results. After protein digestion directly in bis-acrylylcystamine (BAC)-crosslinked polyacrylamide gel, the gel was dissolved and the protein fragments released were analyzed by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS). We found that, compared to other existing methods, the protocol proposed in this study had reproducible peptide recovery and extremely high detection capability for membrane proteins. Moreover, the protein samples stored in the BAC gel were stable for at least one week.
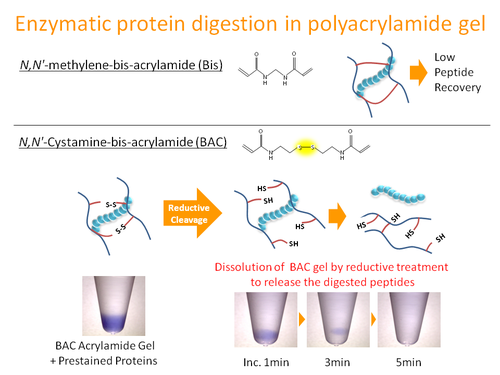
Takemori A. et al., BAC-DROP: Rapid Digestion of Proteome Fractionated via Dissolvable Polyacrylamide Gel Electrophoresis and Its Application to Bottom-Up Proteomics Workflow. J Proteome Res. 2021
Research Focus 2: Quantitative Proteomics
Targeted Transmembrane Proteomics
Isotope dilution mass spectrometry has been widely applied to the quantitative analysis of cellular proteins from small amounts of biological samples. Despite its success for analyzing soluble proteins, accurate quantitation of transmembrane proteins remains challenging because of the lack of suitable internal standards.
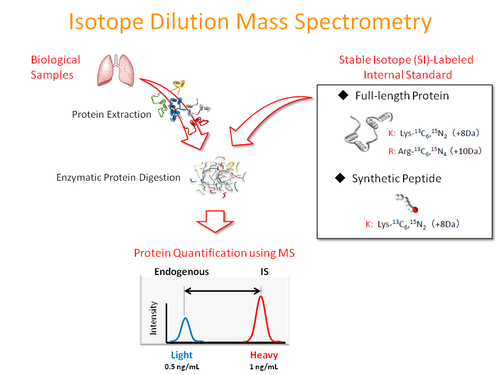
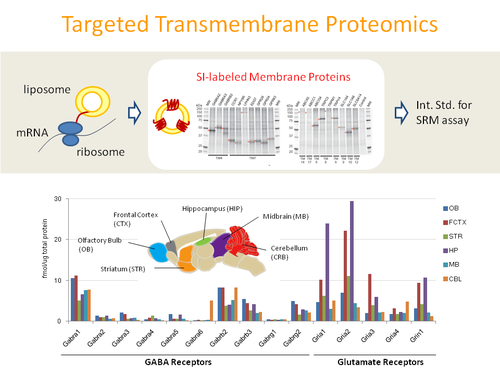
We developed a novel method for the high-throughput synthesis of stable isotope-labeled, full-length transmembrane proteins as high quality internal standards using a wheat germ cell-free protein synthesis system. We synthesized 263 transmembrane proteins by incorporating stable isotope-labeled amino acids in the presence of liposomes. We also established a targeted quantitation assay using the combination of a synthesized internal standard mixture and selected reaction monitoring, and quantified neurotransmitter receptors in different regions of the mouse brain. Our study demonstrates that the wheat cell-free system is powerful in the emerging field of targeted transmembrane proteomics.
Ultra-high throughput biosynthesis of stable isotope labeled standards for global proteome quantification
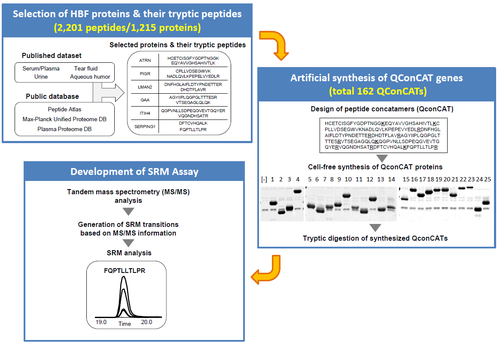
Targeted protein quantification techniques using SRM/MRM generally require the pre-determination of analytical conditions using reliable reference samples. However, comprehensive SRM assay information for the human proteome is currently not available. In this study, we have developed a large-scale SRM assay targeting human body fluid proteins based on previously published proteomic analyses of serum/plasma, tears, aqueous humor, and urine, and synthesized a reference peptide library of 2,201 targeted peptides representing 1,215 human body fluid proteins. Following the liquid chromatography-coupled tandem MS analysis of this library, we successfully developed large-scale SRM assays for human body fluid proteome.